の アンデルセン病 グリコーゲン蓄積症の特に重篤な形態であり、異常なグリコーゲンの形成を特徴とする遺伝性疾患です。病気の予後は非常に悪いです。
アンデルセン病とは?

©ag visuell-stock.adobe.com
のコンテキストで アンデルセン病 異常な形のグリコーゲンが保存されています。このグリコーゲンはアミロペクチンと似た構造をしており、その大部分は植物性デンプンに含まれています。通常、グリコーゲンは高度に分岐しています。しかし、アンデルセン病では、弱く分岐した多糖類しかありません。
この疾患の特徴は、肝臓が急速に肥大し、それがすぐに肝硬変を引き起こすことです。異常な多糖類は分解できなくなり、蓄積し続けます。酵素アミロ-1,4-1,6-トランスグルコシダーゼの欠乏または欠如でさえ、グリコーゲン形成の欠陥の原因となっています。この多糖分子に分岐を提供します。
病気は非常にまれですが、それでもさまざまな形で発生します。非常に深刻な形で、子供はしばしば死産です。後の年齢で始まる軽いフォームも説明されています。しかし、いずれの場合でも、第3染色体にある遺伝子(GBE1)に変異があります。
原因
アンデルセン病の原因は、染色体3の遺伝子GBE1の遺伝的欠陥であり、常染色体劣性形質として遺伝する可能性があります。この遺伝子は、酵素アミロ-1,4-1,6-トランスグルコシダーゼの合成に関与しています。この酵素がないか、機能が限られている場合、通常のグリコーゲンは合成できなくなります。酵素は、多糖分子の分岐を担っています。
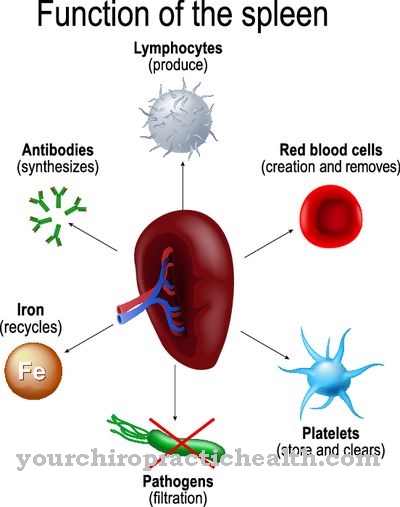
この分岐が行われない場合、または不完全にしか行われない場合は、グリコーゲンが作成されます。グリコーゲンは分解されなくなり、迅速なエネルギー供給ができなくなります。逆に、それは肝臓、脾臓、リンパ節に急速に蓄積します。各食事の後、未使用のグルコースの一部は肝臓に輸送され、予備物質であるグリコーゲンとして保存されます。
ただし、この予備資料は現在の形では使用できません。異常なグリコーゲンの絶え間ない蓄積は、肝臓と脾臓をますます拡大させ、必然的に両方の臓器の破壊につながります。
症状、病気、徴候
アンデルセン病は、異常な変動によって現れます。これは、異常なグリコーゲンが絶え間なく貯蔵されることであり、もはや分解することはできません。しかし、病気の重症度は異なる場合があります。それにもかかわらず、アンデルセン病の予後は全体的に非常に不良です。最も顕著な症状は、常に拡大する肝臓であり、そこから肝硬変が急速に発症します。
最も深刻な形態は、出生前の子供の動きの欠落または減少を通じて現れます。胎児は関節のこわばりと肺低形成の兆候を示しています。通常、これらの場合、子供は死んで生まれます。古典的なケースでは、子供はまだ出産時にまだ発達しています。ただし、人生の最初の数か月で、肝腫大(肝臓の肥大)と筋緊張低下(筋緊張の欠如)が発症します。
全体として、子供の発達は遅れています。病気は急速に進行します。肝臓は肝硬変を発症します。また、門脈圧が上昇し、脾臓が肥大します。肝硬変により、食道内に静脈瘤が発生し、対応する出血と腹水を伴います。通常、死は幼児期に起こります。まれなケースでは、病気は後で始まり、筋力低下と心不全の症状を示します。神経症状もここで発生します。
診断と疾患の経過
診断は臨床像に基づいて行うことができ、臨床検査、肝生検および分子遺伝学的検査を伴うことができます。組織学的検査では、染色可能なアミロペクチン様構造の細胞内蓄積が顕著です。 関与する酵素は、肝細胞、線維芽細胞、白血球で調べられます。アミロ-1,4-1,6-トランスグルコシダーゼの証明された欠乏は診断を裏付ける。
合併症
原則として、子供の寿命はアンデルセン病によって大幅に減少するか、子供が死んで生まれます。これは、特に親族や親に対して、深刻な心理的不満やうつ病を引き起こす可能性があります。ほとんどの場合、それらは心理的治療に依存しています。
影響を受けた子供たちは、肝硬変に苦しんでおり、最終的には死に至ります。さらに、関節も硬くなり、この不満のために動きができなくなります。子供の精神発達もアンデルセン病によって深刻に障害されているため、影響を受ける人は通常、常に他の人の助けに依存しています。心不全や筋力低下が起こることは珍しくありません。
患者は心臓死で死亡することもあります。残念ながら、アンデルセン病は治せません。新しい肝臓への損傷も発生するため、肝臓の移植でも症状が緩和されるのは短時間だけです。これは最終的に子供の死につながります。それまでは、しかし、苦情や症状は医療措置の助けを借りて制限することができます。
いつ医者に行くべきですか?
アンデルセン病は、重症の場合、子宮内で胎児を死に至らしめる遺伝病です。したがって、妊娠中の女性は、妊娠中に異常や異常に気づいたらすぐに治療を受けなければなりません。妊娠中の母親が胎児に問題があるのではないかと漠然と感じている場合は、医師に相談する必要があります。 新生児が出生後の最初の数日および数週間生き残る場合、特異性がさらなる発達の過程で明らかになるとすぐに医師が必要です。筋力低下や運動障害がある場合は、医師の診察を受けてください。
成長障害は、既存の疾患の兆候であり、明確にする必要があります。心臓の異常、体の変形、子供の行動の不一致を調べて治療する必要があります。多くの場合、病気は臓器の肥大を引き起こします。これらの場合、特に肝臓または脾臓が影響を受けます。
したがって、同じ年齢の赤ちゃんや子供と直接比較して上半身の異常な形状が発生するとすぐに医師が必要です。皮膚の変色または皮膚の外観の他の不規則性は、健康障害のさらなる兆候です。黄色がかった顔や目は医師が評価する必要があります。
治療と治療
病気は遺伝性であるため、原因となる治療を行うことはできません。治療は症状のみです。治療の一環として、医師は主に発生する合併症に焦点を当てます。これは門脈回路の圧力を下げます。アルブミンと凝固因子の代用もあります。
肝不全が発生した場合、肝移植は寿命を延ばすことができます。しかし、肝移植をしても治らない。遺伝的欠陥が存在し、新しい肝臓に異常なグリコーゲンの沈着も引き起こします。欠陥のある多糖類の保存は、脾臓およびリンパ節のいわゆる細網組織球系の他の臓器で継続するため、肝移植が成功した後でも深刻な合併症が発生する可能性があります。
網状組織球系は免疫系の一部であり、網状結合組織の細胞を含む。これらの細胞は、それらを分解して体外に運ぶために粒子と物質を蓄えます。しかし、ここでも欠陥のある多糖分子の分解は不可能です。
見通しと予測
アンデルセン病は比較的予後不良です。代謝性疾患はこれまでのところ治癒可能ではなく、深刻な肝障害を引き起こしています。場合によっては、筋肉の不調や付随する疾患が発生し、治療せずに放置すると、徐々に進行します。寿命は状態によって厳しく制限されます。病気の子供は平均して2歳から5歳に達します。初期の肝移植は予後を改善します。特に人生の最初の数か月に肝移植がない場合は特に、この疾患の典型的な形態では、予後は不良です。
原則として、長期予後は疾患の程度、重症度、進行に基づいています。アンデルセン病は最も深刻なグリコーゲノースの1つです。生活の質は通常、肝臓の問題やその他の症状のために大幅に低下します。鎮痛薬と包括的な治療は子供の健康を改善しますが、それらはまたリスクと関連しています。責任のある肝臓専門医が予後を提供します。
寿命は状態によって厳しく制限されます。未検出の疾患で発生する可能性のある随伴性の病気も予後に含まれます。したがって、アンダーセン病は全体として予後不良です。新しい治療方法は、将来的に改善をもたらす可能性があります。
防止
アンデルセン病の予防は、子孫がこの病気を遺伝しないという事実に言及することができるだけです。アンデルセン病は常染色体劣性遺伝で受け継がれるため、遺伝ではいくつかの世代をスキップできます。家族や親戚にすでにアンデルセン病の症例があった場合、人間の遺伝子検査を実施する必要があります。
両方の親で遺伝子が見つかった場合は、遺伝カウンセリングが推奨されます。この場合、子孫は25%の確率でアンデルセン病を発症します。
アフターケア
アンデルセン病は治癒できないため、症状の治療と起こりうる合併症の封じ込めが、治療期間中の主な焦点です。治療の一環として実施される介入後は、フォローアップケアが必要です。肝移植が発生した場合、専門家によるフォローアップケアが肝移植にとって非常に重要です。
手順の後、これは新しい肝臓が体によって拒絶されないことを保証します。特別な薬は体の免疫反応を抑制します。しかしながら、その結果、病原体に対する体の抵抗力が弱まり、それはさらなる治療で考慮に入れられなければなりません。この間、患者は定期的に血液検査を受けなければなりません。副作用として起こり得る拒絶反応または他の深刻な合併症(腎臓機能不全など)がないことを確実にするために注意が払われる。
アンデルセン病の主要な症状は肝移植直後に改善することができますが、欠陥のあるグリコーゲンの沈着が続くため、移植後でも合併症と進行性症状が予想されます。担当の肝臓専門医は、予後と治療の経過についてより詳細な情報を提供できます。
自分でできる
アンデルセン病患者がとれる自助措置は、存在しないものに限られています。病気は遺伝的原因を持ち、対症療法にもかかわらず制御することができないので、影響を受ける人の可能性はすぐに使い果たされます。彼は、担当の医師から与えられた食事療法とライフスタイルのアドバイスを真剣に受け止め、それを実行することをお勧めします。
さらに、肝移植後、影響を受ける人は穏やかな行動を考慮する必要があります。アルコール、脂肪の多い食品、運動は避けてください。これにより、体が新しい臓器を本当に受け入れることが容易になります。ただし、成功したフォローアップケアを含め、移植が成功しても4型グリコーゲン症自体を止めることはできません。
これは常染色体劣性遺伝性疾患であるため(数世代をスキップする可能性があります)、家族を計画するときに遺伝プロファイルを作成することは理にかなっています。アンデルセン病に罹患している人々は彼らの遺伝子についてすでに知っていますが、この点に関する分析は家族にとって特に価値があります。このようにして、適切な家族計画を通じてトリガー遺伝子の伝染を防ぐことができます。しかし、少なくとも、自分の子孫の病気のリスクについて確実性を得ることができます。











.jpg)