の コール酸 脂肪の消化に役割を果たす一次胆汁酸です。それは脂質を乳濁液に安定化させ、それらをリパーゼに対して脆弱にします。コール酸欠乏症では、脂肪の消化が妨げられますが、これは主に便の硬さの変化で顕著です。
コール酸とは何ですか?
コール酸は2つの主要な胆汁酸の1つであり、 12α-トリヒドロキシコラン酸 専用。一次形態の2番目の胆汁酸はケノデオキシコール酸と呼ばれます。体自身の酸生成の出発物質はコレステロールです。生産の中間段階はプレグネノロンです。酸の生合成は肝臓で行われます。コール酸は、肝臓で最も一般的に生成される4つの酸の1つです。
その乳化特性のため、ステロイドはコレステロール代謝において主要な役割を果たします。酸は生合成で胆汁酸塩に変換され、コール酸塩を形成します。二次形態では、コール酸はデオキシコール酸を生成します。コール酸を使用して胆石を溶解し、肝臓の健康をサポートします。コール酸は腸肝循環に関与し、10回以上リサイクルされています。
解剖学と構造
コール酸は無色の結晶性物質で、苦味があり、融点は摂氏198度です。体自身の酸の化学式はC24H40O5です。この物質は水に溶けにくいだけです。このようにして、実際に混和しない物質を混合し、これらのエマルションを安定させるのに役立ちます。コール酸はステロールのグループからのステロイドであり、脂質のクラスに分類されます。それらの分子は親油性グループを形成します。
つまり、油脂を簡単に溶解することができます。脂質は水に不溶です。それらは、4つのトランスリンクされた炭素環で構成される基本構造を持っています。脂肪消化におけるすべての胆汁酸は、疎水性部分と親水性部分で構成されています。したがって、脂肪の周りを閉じることができるため、脂質が胃腸管に吸収されます。この文脈において、コール酸はコレステロールの吸収に特に不可欠です。
機能とタスク
消化中、脂肪消化酵素リパーゼはコレステロールエステルなどの脂質から遊離脂肪酸を分解します。リパーゼは体内の貯蔵脂肪を利用できるようにし、脂肪の利用にも関与しています。遊離脂肪酸を分離しないと、脂質は体にとって消化されにくく、腸壁に吸収されません。遊離脂肪酸の分離がより効果的になるように、消化管内の脂質が安定化して乳濁液を形成します。
コール酸などの胆汁酸は、このプロセスで乳化剤として機能します。このようにして、脂肪がリパーゼにアクセスしやすくなります。これは、肝臓でのコール酸の合成が先行します。ここで、コール酸はグリココール酸またはグリシンコール酸アミドおよびタウロコール酸またはタウリンコール酸アミドに変換されます。これらの酸は胆汁に輸送されます。それらは塩として消化器系に入ります。コール酸が乳化剤としての機能を果たすと、小腸がそれらを再び吸収します。酸の90%以上は、Na +共輸送を通じて二次的な活性型で吸収されます。
約2パーセントは、小腸および大腸での非イオン性およびイオン性の拡散によって受動的に再吸収されます。これらの吸収プロセスを通じて、コール酸の約3%のみが大腸に放出されます。サイトゾル輸送タンパク質は、コール酸の大部分を陰イオン交換体とともに基底外側膜を介して門脈の血液に輸送します。
このようにして、コール酸は肝臓に戻ります。それらは、臓器の肝細胞に結合されており、再び体に利用できます。コール酸のほんの一部のみが毎日便から失われます。これらの損失を補うために、肝臓は毎日少量のコール酸を再合成します。
病気
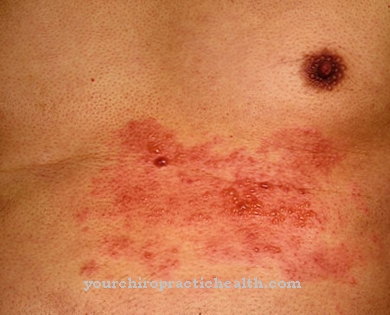
コレステロールがコール酸乳濁液から抜け出すと、胆石が形成されます。したがって、胆石症は、コール酸の機能不全の兆候です。この物質はもはやこの方法で脂肪を消化する機能を果たすことができないため、コール酸の欠乏も胆石を引き起こす可能性があります。コール酸欠乏症にはさまざまな原因があります。先天性胆汁酸合成の欠陥が存在する可能性があります。
小腸の炎症部位からコール酸が吸収されなくなるため、慢性的な腸の炎症があっても、コール酸は十分ではなくなります。コール酸が毎日大量に大腸に移動し、それによって便に排泄される場合、小腸と大腸を分離しているフラップが炎症や腫瘍によって影響を受ける可能性があります。原因が慢性的な腸の炎症である場合、主な疾患は自己免疫クローン病である可能性があります。肝疾患もコール酸欠乏症を引き起こす可能性が高いです。
たとえば、肝臓で十分なコール酸が合成されない場合、1日の損失は、長期的には便で十分に補うことができません。 1日あたりの損失はごくわずかですが、長期的に見れば、合計されて一般的なコール酸欠乏症を引き起こす可能性があります。このような欠乏は通常、便の粘り強さの変化で顕著になります。特に、消化管内の脂肪が十分に吸着されず、排泄されるため、脂肪便はコール酸の欠乏を示します。




.jpg)

.jpg)